导读来自科学院上海高级研究所,浙江大学和丹麦技术大学的国际合研究小组报告了一种在催化反应过程中以原子精度操纵界面结构的原位策略。研究结
来自科学院上海高级研究所,浙江大学和丹麦技术大学的国际合研究小组报告了一种在催化反应过程中以原子精度操纵界面结构的原位策略。研究结果发表在最新一期的《科学》杂志上。
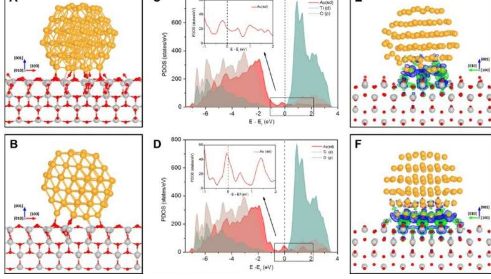
纳米颗粒和底物之间的界面在非均相催化中起着至关重要的作用,因为大多数活性位点都位于界面的周围。通常认为该接口是固定的且不可更改的,因此很难在反应性环境中进行调整。结果,通过精确控制界面结构来促进催化活性一直是挑战性的。
在这项研究中,科学家首先使用环境透射电子显微镜直接观察了原子级CO氧化过程中金纳米颗粒在二氧化钛(TiO 2)表面上的外延旋转。在O 2环境下实时观察到Au纳米颗粒与TiO 2(001)表面之间具有完美的外延关系。
然后进行了包括密度泛函理论计算和热力学分析在内的理论计算,表明外延取向可以通过改变周边界面处的O 2吸附覆盖率来诱导。Au纳米颗粒在Au-TiO 2界面处吸附更多的O 2分子时更稳定,但随着CO 2消耗O 2而变得较不稳定。
为了利用Au-TiO 2界面的活性增强,研究人员进行了另外的俯视观察,发现当在CO和O 2反应性环境中从500°C冷却至20°C时,该构型保持不变,从而显示了Au的旋转纳米颗粒在反应条件下也与温度有关。
利用Au纳米粒子可逆和可控的旋转,科学家通过改变气体和温度实现了原子级活性Au-TiO 2界面的原位控制。
这项研究揭示了在原子尺度上反应条件下催化界面结构的实时操纵,这可能会激发未来在操作条件下实时设计催化界面的方法。