准确知道蛋白质在哪里受挫可能对制造更好的药物有很大帮助。这是莱斯大学科学家进行的一项新研究的结果,该研究正在寻找稳定或破坏生物分子关键部分的机制。
赖斯理论家彼得·沃里斯(Peter Wolynes),第一作者和校友明辰晨(Mingchen Chen)及其同事在理论生物物理中心的原子尺度模型表明,不仅蛋白质中某些特定的受挫序列对于使它们发挥功能是必需的,找到它们还为我们提供了线索。达到更好的药物特异性。
Wolynes说,这些知识也可以帮助设计副作用更少的药物。
该小组的开放获取研究发表在《自然通讯》上。
原子尺度模型将可能的结合位点内的相互作用归零,而不是指导其折叠的蛋白质中的绝大多数相互作用。较精细的分辨率模型允许并入诸如化学活性配体等辅助因子,包括药物分子。研究人员说,这种能力为为什么配体只能被特定的蛋白质而不是其他蛋白质最好地捕获提供了新的见解。
Wolynes说,又名药物,“非天然配体”倾向于与蛋白质中那些受挫的口袋结合得最好,一旦药物结合,这些口袋中的受挫感就会最小化。有一种方法来查找然后学习这些受挫最少的站点的详细信息,将有助于制药公司消除大量的试验和错误。
Wolynes说:“进行药物设计的标准方法是在蛋白质上尝试10,000个结合位点以找到合适的结合位点。” “我们是说您不必对所有可能的结合位点进行采样,只需一个合理的数字即可了解在本地环境中可能起作用的统计信息。
他说:“这是进行民意测验和实际进行选举之间的区别。” “民意调查比较便宜,但是您仍然需要检查一下。”
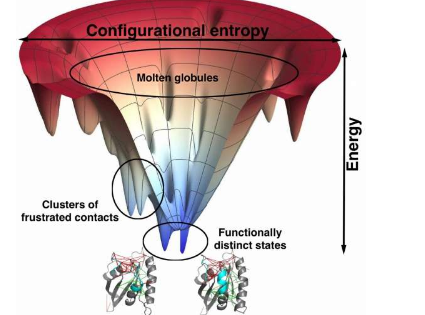
赖斯大学的研究人员以蛋白质如何折叠的能量图论而闻名。它通常采用粗粒模型,其中氨基酸仅由几个位点表示。
与尝试确定每个残基中每个原子随时间的位置相比,该策略所需的计算能力更低,但事实证明,该策略可根据其序列预测蛋白质的折叠方式非常准确。但是对于这项研究,研究人员在原子水平上对蛋白质和蛋白质-配体复合物进行了建模,以了解它们是否能够发现挫折感如何使蛋白质的某些部分具有与其他分子结合所需的灵活性。
Wolynes说:“以全原子分辨率进行建模的一大优点是,它使我们能够评估药物分子是否很好地适合结合位点。” “这种方法能够快速显示出某种药物的结合位点将受到最小程度的抑制还是保留在一个受抑制的区域。如果分子结合后该位点仍然受到抑制,则蛋白质可能会重新排列,或者药物可能会在这种情况下改变其方向一种可能引起副作用的方式。”
对受挫的位点进行建模(有时进行更改以查看会发生什么情况),让研究人员了解药物特异性与结合口袋的关系。他们写道,挫败感分析提供了“一种筛选用于药物发现的更具体化合物的途径”。
沃林斯说:“这种沮丧的观念出现在我们蛋白质折叠研究的开始之初。” “当我们将其应用于真实的蛋白质分子时,我们发现了一些折叠机制违反了我们根据理想漏斗预测的例子。然后,我们发现这些与漏斗图像的偏差实际上是在蛋白质受挫的情况下发生的。
他说:“就像证明规则的例外一样。” “一直以来都是正确的事情可能是微不足道的。但是,如果不是只有1%的时间是正确的,那么这是一个有待解决的问题,我们已经能够使用我们的结构预测软件AWSEM做到这一点。”
正如该小组在另一篇论文中所描述的那样,可以扩展该软件以在原子水平上分析挫败感。但是,跟踪蛋白质中每个原子的计算成本很高,以至于研究人员需要一种方法来采样特定区域的运动,在这些区域中,挫折可能会使折叠路径混乱。
Wolynes说:“ Mingchen意识到有一个有效的算法可以在结合位点对局部环境进行采样,但保持原子分辨率。”他指出,他和Chen(现为私营企业)正在使用该模型研究可能的治疗方法,包括相关药物。